通过计算发现新材料虽然可以简化实验合成前的筛选过程,但因材料组分和结构的巨大潜在组合,这一筛选过程仍然道阻且长,系统地探索材料组分和结构空间亦面临挑战。
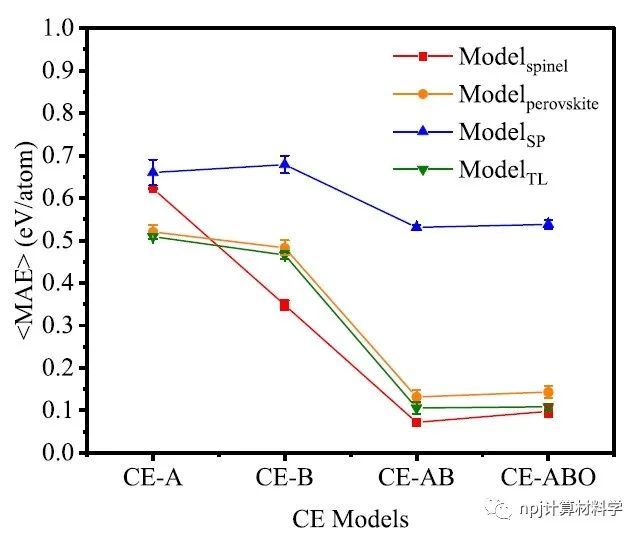
Fig. 1 Performance (<MAE>) of prediction of various test datasets using the CE feature models with different center atom definitions.
如果目标材料的数据有限则困难更大,此时从其他材料已知的大型数据集中作跨晶体结构的迁移机器学习,便成为材料设计中的重要策略之一。
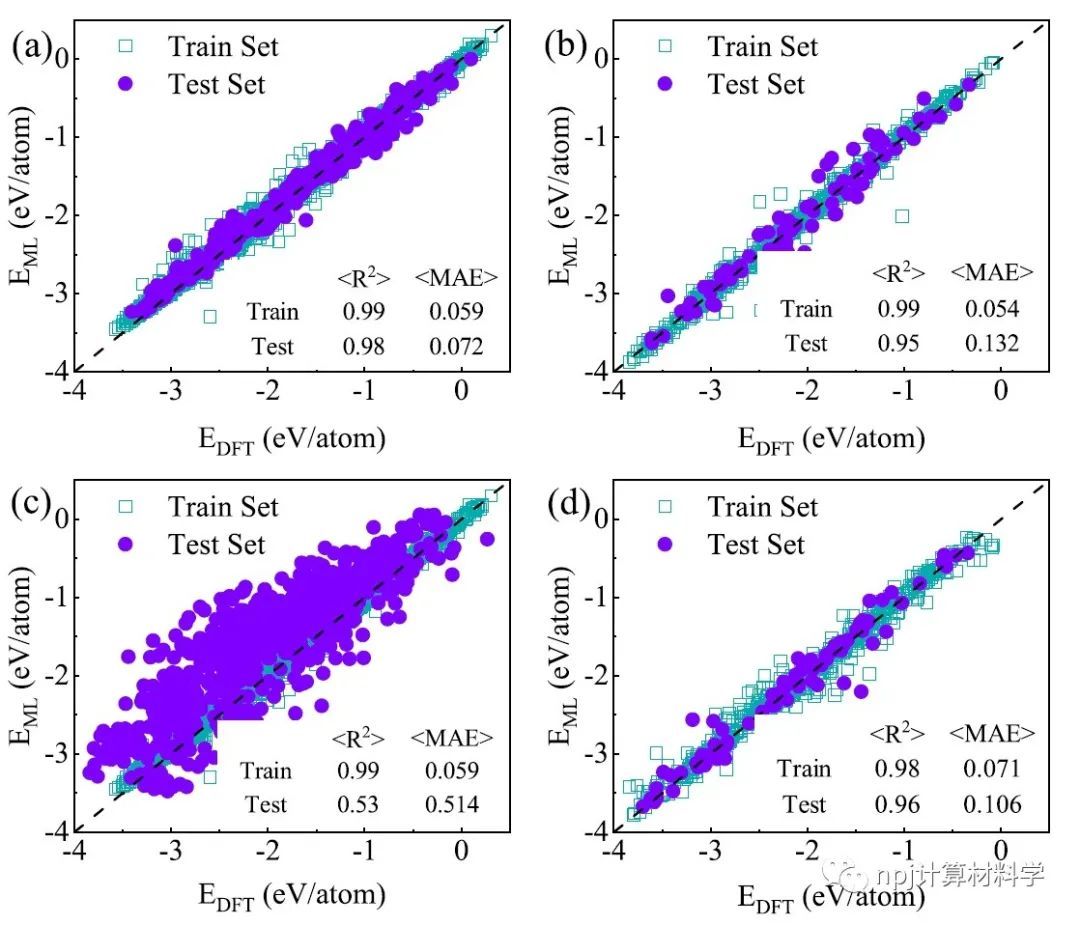
Fig. 2 Formation energies predicted by the ML with CE features (DNN-CE) and DFT using various datasets.
来自上海大学材料基因组工程研究院的刘轶教授和冯凌燕教授团队,提出了一种基于大规模尖晶石氧化物计算数据集的深度迁移学习方法,用于预测热力学稳定的钙钛矿氧化物。
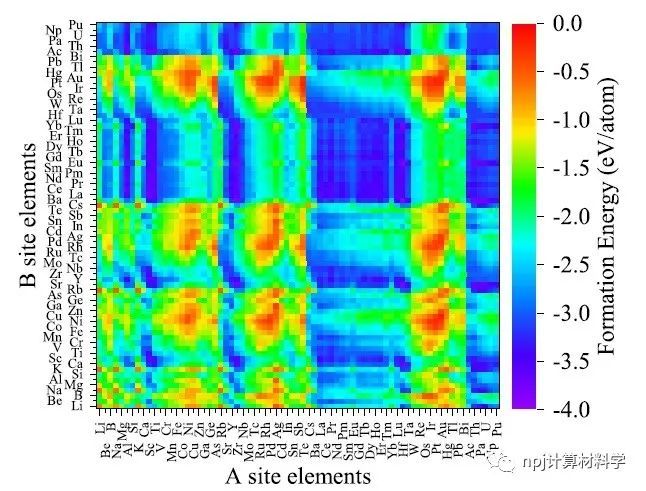
Fig. 3 Heat map of formation energies of 5329 ABO3 perovskite oxide structures predicted by the transferred learning model in this work, containing 73 constitution elements at the A and B sites, respectively, sorted by the atom number.
他们使用5329个尖晶石氧化物结构的计算形成能,开发了一个具有“中心–环境(CE)”特征的深度神经元网络(DNN)源域模型,然后通过学习855个钙钛矿氧化物结构的小数据集对DNN模型参数进行微调,获得了在目标域钙钛矿氧化物中具有良好可迁移性的迁移学习模型。

Fig. 4 Heat map of tolerance factor of 5329 perovskite oxide structures calculated in this work, containing 73 constitution elements at the A and B sites, sorted by the atom number.
通过迁移学习模型预测的钙钛矿结构的形成能平均绝对误差(MAE)仅为0.106 eV/atom,优于仅使用钙钛矿数据训练模型的0.132 eV/atom。基于迁移学习模型,作者快速预测了包含73种元素的5329个潜在钙钛矿结构的形成能。
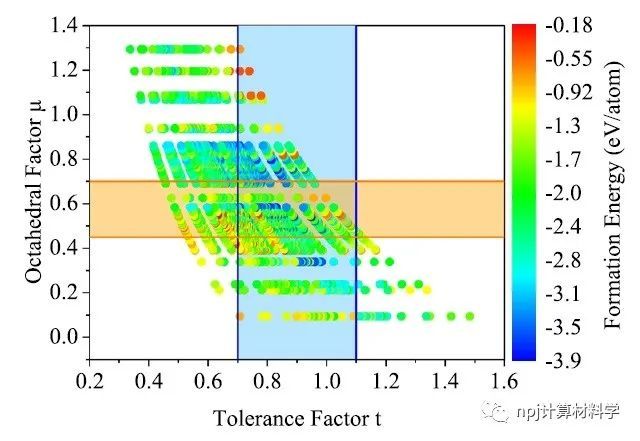
Fig. 5 Tolerance factor (t) vs. octahedral factor (μ) scatter plot ofperovskite oxide structures, where the colormap corresponds tothe transfer learning predicted formation energy of perovskitestructure.
结合预测的形成能和包括容忍因子(0.7 < t ≤ 1.1)和八面体因子(0.45 < μ < 0.7)的结构因子标准,他们又预测了1314种潜在的热力学稳定的钙钛矿氧化物。在这预测的1314种潜在钙钛矿氧化物中,已被实验合成证实的有144种,已被其他计算所预测的有10种,Materials Project数据库中已记录的有301种,其余859种氧化物尚未在文献中报道。

该研究将基于结构信息的机器学习特征和迁移学习方法结合,利用丰富的已知结构数据,花费较低的额外计算成本即可预测新结构,为昂贵的高通量计算筛选材料设计,提供了新的有效的加速策略。

Fig. 7 Crystal structures and constituent elements of spinel oxides and perovskite oxides studied in this work.
预测的新型钙钛矿氧化物为实验合成和探索可再生能源及电子材料应用提供了丰富的候选材料。相关论文近期发布于npj Computational Materials 9:106 (2023)。手机阅读原文,请点击本文底部左下角“阅读原文”,进入后亦可下载全文PDF文件。
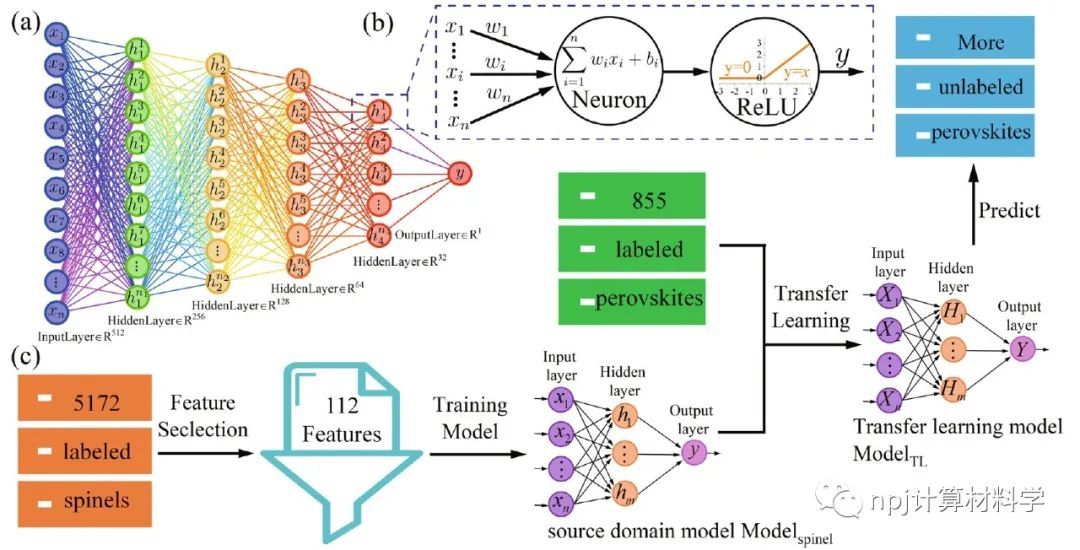
Fig. 8 General schematic diagram of DNN-CE models and the workflow of transfer learning method in this work.
Editorial Summary
Transfer learning across crystal structures: “Center-Environment” feature accelerates materials predicting
Discovering new materials through computational methods has simplified the screening process before experimental synthesis. However, systematically exploring the material space remains challenging due to the vast potential combinations of material compositions and structures. In cases where data on the target material is limited, cross-crystal structure transfer learning from large-scale known datasets of other materials has become an important strategy in materials design.
This study proposes a deep transfer learning approach based on a large-scale dataset of spinel oxide compounds to predict the thermodynamically stable perovskite oxides. Prof. Liu and Prof. Feng’s team at Materials Genome Institute of Shanghai University utilized the formation energy of 5,329 spinel oxide structures to develop a deep neural network (DNN) source domain model with “Center-Environment” (CE) features. The CE-DNN model was then fine-tuned using a small dataset of 855 perovskite oxide structures to achieve a transferable learning model with good performance in the perovskite oxide target domain.
The mean absolute error (MAE) of the perovskite structure formation energy predicted by the transfer learning model is 0.106 eV/atom, which is better than the MAE of 0.132 eV/atom of the model trained solely using small perovskite data. Based on the transfer learning model, the formation energy of 5,329 potential perovskite structures containing 73 different elements was further predicted. Combining the predicted formation energies with structural factor criteria, including tolerance factor (0.7 < t ≤ 1.1) and octahedral factor (0.45 < μ < 0.7), a total of 1,314 potentially thermodynamically stable perovskite oxides were predicted.
Among 1,314 predicted potential perovskite oxides, 144 were experimentally synthesized, 10 were predicted by other computational works, and 301 are documented in the Materials Project database, while the remaining 859 oxides have not been reported in literatures. The combination of structural information features and transfer learning methods in this study enables the low-cost prediction of new structures using existing big data, providing an effective acceleration strategy for expensive high-throughput computational material design. The predicted stable novel perovskite oxides offer a rich platform for exploring novel perovskite experimental synthesis, renewable energy and electronic materials applications.This article was recently published in npj Computational Materials9: 106 (2023).
原文Abstract及其翻译
Center-environment deep transfer machine learning across crystal structures: from spinel oxides to perovskite oxides (跨晶体结构的深度迁移机器学习:从尖晶石氧化物到钙钛矿氧化物)
Yihang Li, Ruijie Zhu, Yuanqing Wang, Lingyan Feng & Yi Liu
AbstractIn data-driven materials design where the target materials have limited data, the transfer machine learning from large known source materials, becomes a demanding strategy especially across different crystal structures. In this work, we proposed a deep transfer learning approach to predict thermodynamically stable perovskite oxides based on a large computational dataset of spinel oxides. The deep neural network (DNN) source domain model with “Center-Environment” (CE) features was first developed using the formation energy of 5329 spinel oxide structures and then was fine-tuned by learning a small dataset of 855 perovskite oxide structures, leading to a transfer learning model with good transferability in the target domain of perovskite oxides. Based on the transferred model, we further predicted the formation energy of potential 5329 perovskite structures with combination of 73 elements. Combining the criteria of formation energy and structure factors including tolerance factor (0.7 < t ≤ 1.1) and octahedron factor (0.45 < μ < 0.7), we predicted 1314 thermodynamically stable perovskite oxides, among which 144 oxides were reported to be synthesized experimentally, 10 oxides were predicted computationally by other literatures, 301 oxides were recorded in the Materials Project database, and 859 oxides have been first reported. Combing with the structure-informed features the transfer machine learning approach in this work takes the advantage of existing data to predict new structures at a lower cost, providing an effective acceleration strategy for the expensive high-throughput computational screening in materials design. The predicted stable novel perovskite oxides serve as a rich platform for exploring potential renewable energy and electronic materials applications.
摘要
在数据驱动的材料设计中,当目标材料的数据有限时,基于大量已知的源材料数据进行迁移机器学习,尤其是还能跨越不同的晶体结构,成为一种有实际需求和应用场景的研究策略。本论文提出了一种深度迁移学习方法,基于已知的大规模的尖晶石氧化物计算数据和新增的少量钙钛矿氧化物计算数据,跨晶体结构预测热力学稳定的新型钙钛矿氧化物。
首先,利用5329个尖晶石氧化物结构的计算形成能,开发了基于包含结构信息的“中心–环境”(CE)特征的深度神经网络(DNN)源域模型,然后通过学习855个钙钛矿氧化物结构的小数据集对DNN模型参数进行微调,获得具有良好可迁移性的针对目标域钙钛矿氧化物的迁移学习模型。基于CE-DNN迁移学习模型,我们进一步预测了包含73种元素组合的5329个钙钛矿结构的形成能。结合预测的形成能和结构因子(包括容忍因子(0.7 < t ≤ 1.1)和八面体因子(0.45 < μ < 0.7))的判据,我们共预测了1314个潜在的热力学稳定的钙钛矿氧化物,其中144个氧化物已被实验合成,其他计算文献预测了10个氧化物,301个氧化物记录在Materials Project数据库中,而另外859个氧化物是本文首次报道。
基于包含结构信息的特征工程,本研究中的迁移机器学习方法利用现有已知的丰富数据,以较低的额外计算成本预测新晶体结构性质,为昂贵的高通量计算筛选提供了有效的加速策略。本工作预测的新型钙钛矿氧化物为探索可再生能源和电子材料应用提供了丰富的材料候选和改进平台。
